

MO/MC法の並列化
計算方法の詳細
-
MO/MC法
- 分子軌道(MO)計算を用いたモンテカルロ(MC)シミュレーション
- 従来のL-J+Coulombicポテンシャルではなく、MO計算(半経験的、非経験的、DFT)によりエネルギーを計算する
- 溶媒和エネルギーを量子化学的に計算することが可能(QM/MM/MC法などと違い、full quantum)
- 数十~数百万回のMO計算を必要とするため、計算時間が膨大であり、その短縮が課題
- 現在は主に、比較的計算時間の短いPM3,AM1法を用いている。
- サンプリング方法
⇒メトロポリスの方法 - 計算モデル
⇒第一溶媒和圏を包む液滴モデル、NPTアンサンブル - 溶媒和エネルギーの算出
⇒溶媒-溶質クラスタ(ΔEaq)及び、溶媒クラスタ(ΔEH2O)の2つエネルギーをステップ毎に計算
⇒ΔEaq よりΔEH2Oを減算することで、溶媒和エネルギー(ΔEsol)を算出する
⇒界面効果、溶媒構造に起因するエネルギー差の排除
- 熱力学的諸量を計算する式
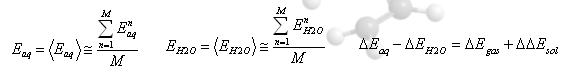
PowerMCプログラム(MO/MC法のインプリメンテーション)
- 開発言語
⇒Fortran90, C++, MPI2 - MO計算ホストプログラム
⇒Gaussian03(for ab initio)
⇒ MOPAC2000(for semiempirical),
⇒GAMESS(for ab initio, 現在開発中) - 擬似乱数
⇒Mersenne Twister Algorithm - コンパイラとライブラリ
⇒Intel Fortran Compiler 8 + MKL7.2 - 計算時間
⇒PM3法、200万ステップ、121原子(PowerMC+MOPAC2000)
⇒約1,370,000秒(Intel(R) Pentium(R) D 3.2GHz 1CPU) =16日間
並列化アルゴリズム
-
MO/MC法高速化の検討
- CPU性能の高い計算機を用いる(Not cluster)
⇒技術的、経済的な側面から限界がある。
⇒SMP、クラスタ(並列計算機)では意味を成さない。 - エネルギー(MO)計算を並列化する
⇒並列化されていないMO計算では高速化できない。
⇒計算ホストにGaussian, GAMESSを用いた場合は可能。 - シミュレーション(サンプリング)を並列化する
⇒メトロポリスサンプリングは、次ステップの計算に、前ステップの計算結果を必要とするリ⇒ニアな計算法であるため、並列化ができなかった。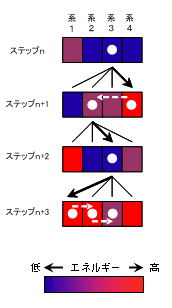
⇒LJポテンシャルを用いる従来のMC法では、計算時間が短いため、並列化の必要はなかった。
⇒MO/MC法において、サンプリング法自体が並列化できれば、大幅な時間短縮になると予想される。
-
メトロポリスサンプリング並列化アルゴリズム
- 系を並列に発生させ、並列にエネルギーを計算
1.初期構造となる系を入力
2.複数台のコンピュータを用いて、並列に系を発生
3.発生させた各系に対し、エネルギーを算出
4.乱数をもちいて、系を一つ選択
5.ΔEを計算し、メトロポリス判定で系を受入or破棄
6.破棄された場合は、別な系を選択し5を再実行。すべての系が破棄された場合は2より再実行.
7.選択された系を次の系として利用し、選択されなかった系は破棄する.
並列化効率
- 受入確率Racc、並列化プロセッサ数N、並列化効率Iparallelの時は以下のように計算される.
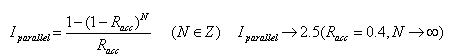
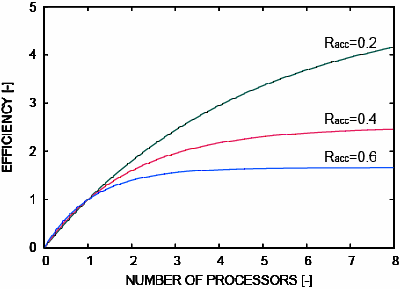
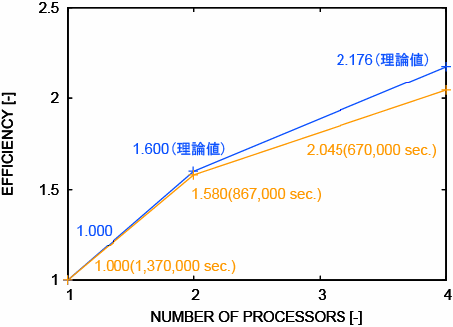
- 一般的なMC計算で適当とされているRacc=0.4のとき、4並列では、Iparallel=2.176となり、約2倍に高速化される。
- 並列化効率は決して良くないが、MO計算の並列化と組み合わせることで、さらなる高速化が期待できる。
並列化による高速化結果
- PM3法、200万ステップ、121原子(Racc=0.40)
- Intel(R) Pentium(R) D 3.2GHz 1 - 4CPU
- ほぼ理論値通りの高速化が実現されている。