量子化学(QM)計算と分子動力学(MD)計算を併用した有機電解質の誘電率と粘度の計算
近年、スマートフォンなどのユビキタス端末の普及により、二次電池の需要が増加しています。新しい電池の開発においては、適切な誘電率と粘度を有する有機電解質の選定が必要となります。 今回は、量子化学(QM)計算と分子動力学(MD)シミュレーションを組み合わせることで、17種類の有機電解質の物性の予測を行いました。
1. 計算方法
1.1. 誘電率εの計算方法
誘電率εは以下のKirkwood-Onsagerの式から求めることができます。
ここで、 は誘電率、 は屈折率、 は分子量、 は系の密度、 は双極子モーメント、 は絶対温度、 はアボガドロ定数、 はボルツマン定数、 は真空の誘電率です。屈折率 はローレンツ-ローレンツ方程式から導かれます。
ここで、 は分子の偏光性です。 また、 はKirkwoodのg因子と呼ばれる値で、溶媒分子の配向性と幾何的配置を反映した相関係数です。水のような極性が強く水素結合ネットワークを形成しやすい分子は実質的に双極子モーメントの大きな一個の分子として振る舞ってしまうため、このg因子で補正しなければ誘電率を過小評価してしまいます。このg因子はMDシミュレーションのトラジェクトリから得られる分子間の距離 と角度 を集計して求められます。ここでrは質量加重座標での分子の重力中心間の距離、 はある分子の中心から半径 の球面内の分子の数、 は分子 と分子 の間の双極子モーメントの角度です。
しかし、上記の式で求められたg因子には双極子モーメント間の長距離静電相互作用が含まれているため、セルサイズや分子数、境界条件などに大きく依存してしまいます。これを解決するため、Zhangらの開発した手法を用いました。電場をゼロにした条件(シ ミュレーション)および電束密度をゼロにした条件( シミュレーション)で求められたg因子を結合させることで、長距離の寄与を相殺して短距離のg因子を計算することができます。
したがって、量子化学計算により双極子モーメント および偏光性 を、MDシミュレーションによりKirkwoodのg因子 および系の密度 を求めることで、誘電率 を算出することができます。
QM計算のみでも誘電率の算出は可能ですが、極性溶媒についてもより良い精度を確保するためにMDシミュレーションで得られる分子間相互作用を反映したKirkwoodのg因子を導入することが望ましいと考えられます。
せん断粘度ηの計算方法
せん断密度 は、トラジェクトリから速度自己相関関数を用いて、グリーン-久保の関係式から以下のように求めることができます。
2. 結果と考察
下図に示した17種類の有機電解質についてQM計算を用いてB3LYP/6-311+G(d,p)レベルで双極子モーメントμと偏光性αを算出しました。
また、MDシミュレーションは、一辺が40~45Åの立方体セルに各分子を512個ランダムに配置した構造を初期構造とし、NPTアンサンブルを用いて系の密度が十分に平衡状態に至る(10ns)までMDを実行しました。温度と圧力の制御にはそれぞれNose-Hoover法とBerendsen法を用いました。力場はGeneral Amber Force Field(GAFF)を適用しました。
g因子Gについては10nsのうち、8nsから10nsまでのトラジェクトリを用いて算出しました。せん断粘度については、平衡状態に至った最終構造から99個の異なる初期構造を作成し、それぞれの構造においてNVEアンサンブルを用いてMDシミュレーションを実行し統計平均を算出しました。
-
計算対象とした17種類の有機電解質
-
誘電率と粘度の計算値と実測値
| 化合物 | 計算値 誘電率 [-] | 計算値 せん断粘度 [mPa・s] | 実測値 誘電率 [-] | 実測値 せん断粘度 [mPa・s] |
|---|---|---|---|---|
| AN(acetnitrile) | 36.98 | 0.3168 | 35.85 | 0.35 |
| BC(Butylene carbonate) | 59.34 | 2.8978 | 58 | 3.1 |
| DEC(Diethyl carbonate) | 2.81 | 1.0342 | 2.82 | 0.75 |
| DEE(1,2-Diethoxyethane) | 5.93 | 0.6111 | 7.2 | 0.602 |
| DG(Diethylene glycol dimethyl ether) | 11.60 | 1.2211 | 7.23 | 1.003 |
| DMC(Dimethyl carbonate) | 3.43 | 0.7910 | 3.12 | 0.63 |
| DME(1,2-Dimethoxyethane) | 5.98 | 0.5545 | 7.3 | 0.356 |
| DOx(1,3-Dioxolane) | 11.73 | 0.8322 | 13.6 | 0.531 |
| AC(Ethylene carbonate) | 86.10 | 2.1019 | 90.5 | 1.9 |
| EMC(Ethyl methyl carbonate) | 3.05 | 0.9386 | 2.93 | 0.68 |
| GBL(g-Butyrolactone) | 48.49 | 1.3997 | 41.78 | 1.722 |
| GVL(g-Valerolactone) | 41.07 | 1.8832 | 34.47 | 2.0 |
| NMP(N-Methyl-2-pyrrolidinone) | 31.95 | 1.5568 | 32 | 1.666 |
| PC(Propylene carbonate) | 69.28 | 2.7554 | 64.97 | 2.501 |
| PE(Ethyl propionate) | 5.90 | 0.8253 | 5.717 | 0.575 |
| TG(Triethylene Glycol Dimethyl Ether) | 6.44 | 2.6192 | 7.62 | 1.96 |
| THF(Tetrahydrofuran) | 8.83 | 0.4159 | 7.51 | 0.45 |
-
NPT平衡化後のスナップショット

-
誘電率と粘度の計算値と実測値との相関
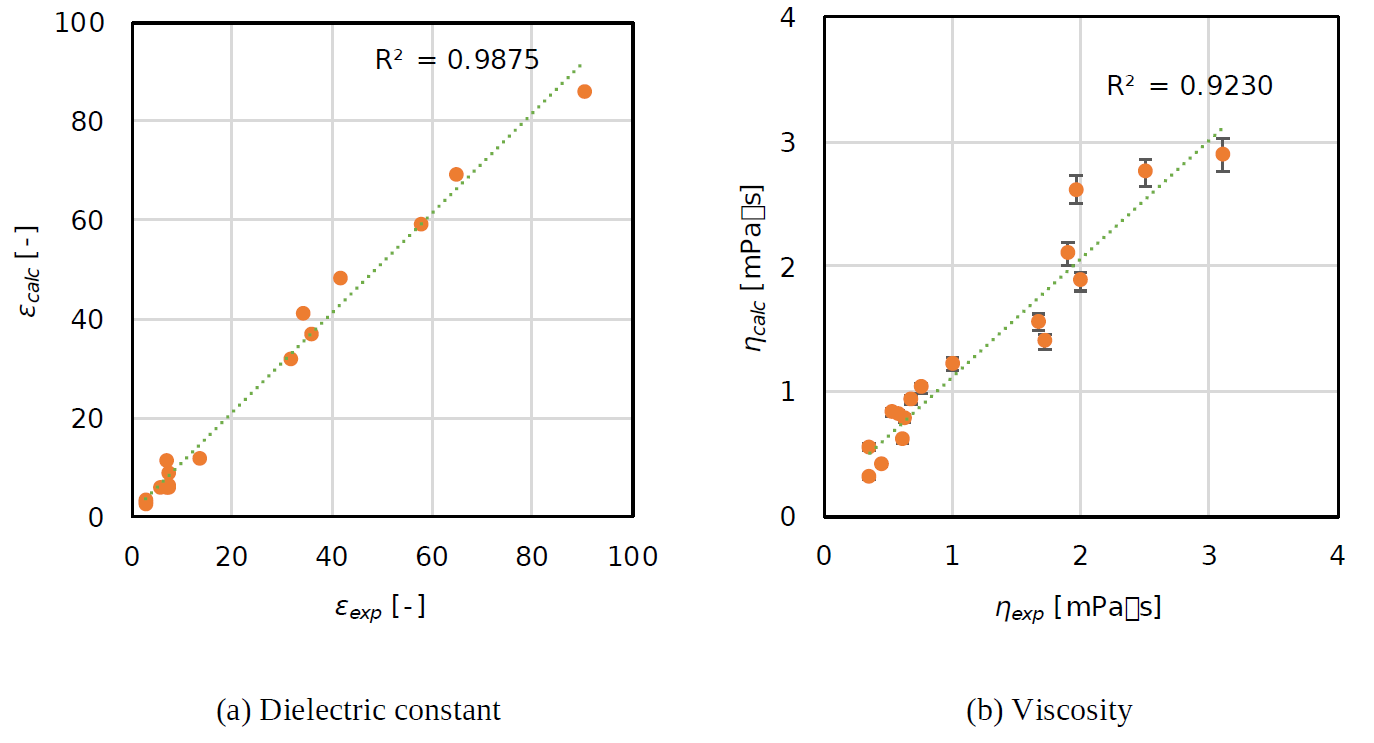
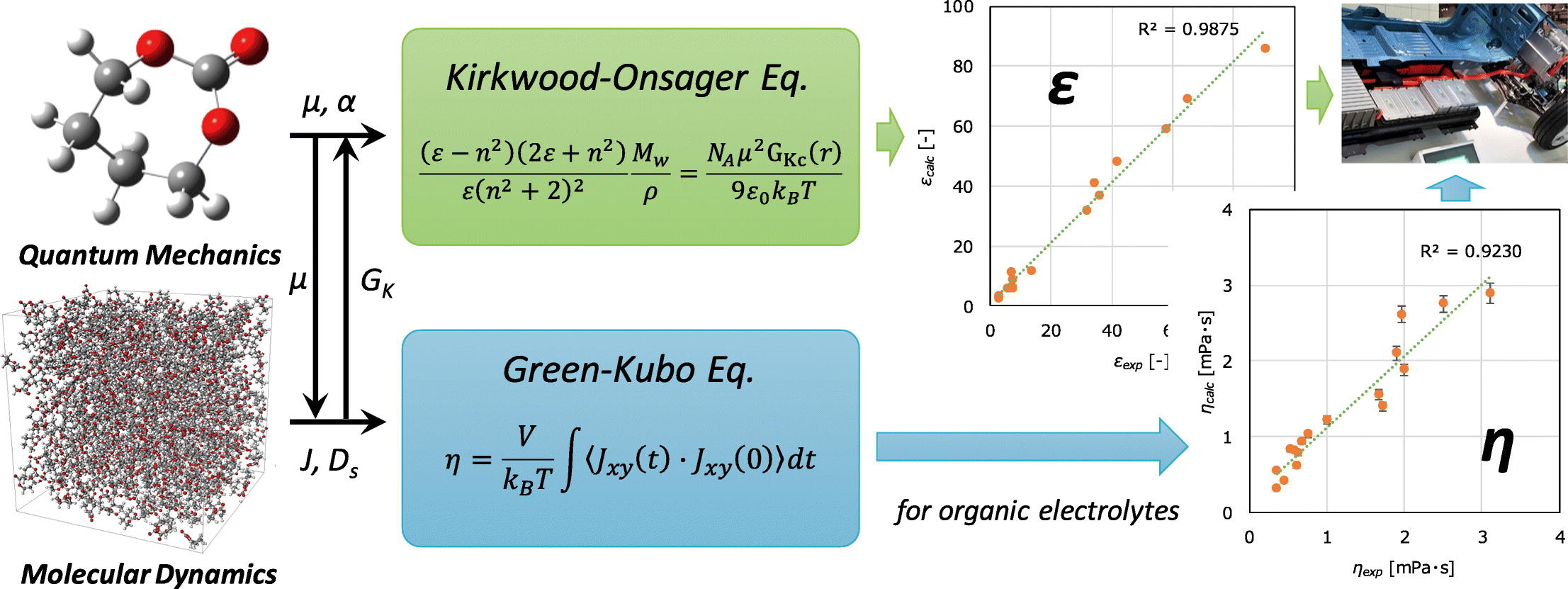
得られた誘電率 とせん断粘度 の計算値と実測値の相関図を示しました。誘電率の相関係数()は0.9875と算出されました。誘電率は様々な種類の有機電解質においても実測値と非常に良い相関があると言えます。粘度の相関係数()は0.9230であり、こちらも実測値と良い一致が見られました。
- g因子の有無による誘電率の精度向上
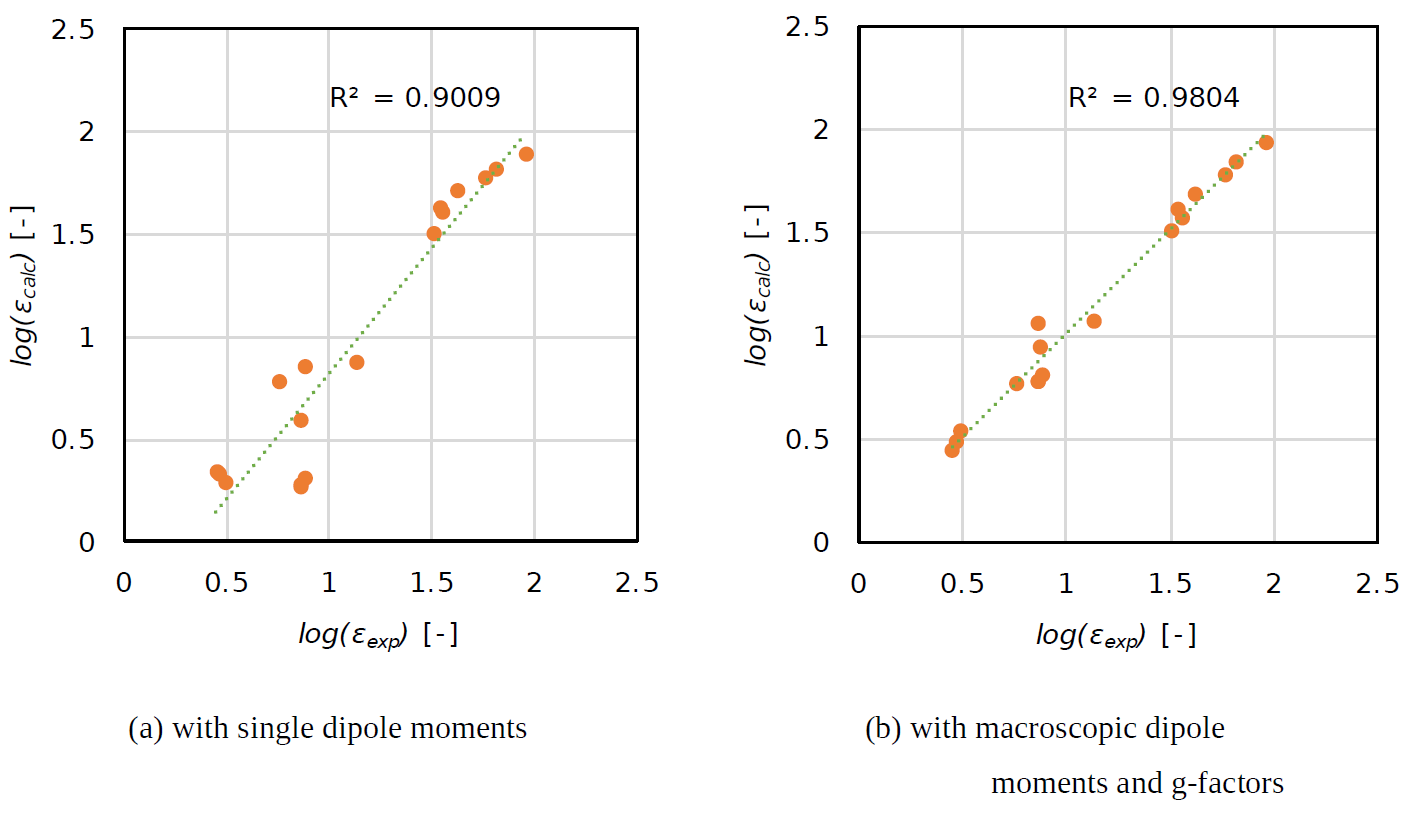
上図の(a)はg因子を考慮せずに1分子の双極子モーメントにより計算した( とした場合の)log の計算値と実測値の相関図であり、(b)はMDシミュレーションで求めたg因子を考慮したlog の計算値と実測値の相関図です。
(a)は低誘電率領域において相関係数()が0.9009であったことに対し、(b)は相関係数()が0.9804と有意な改善が見られました。
- O-H間距離の動径分布関数
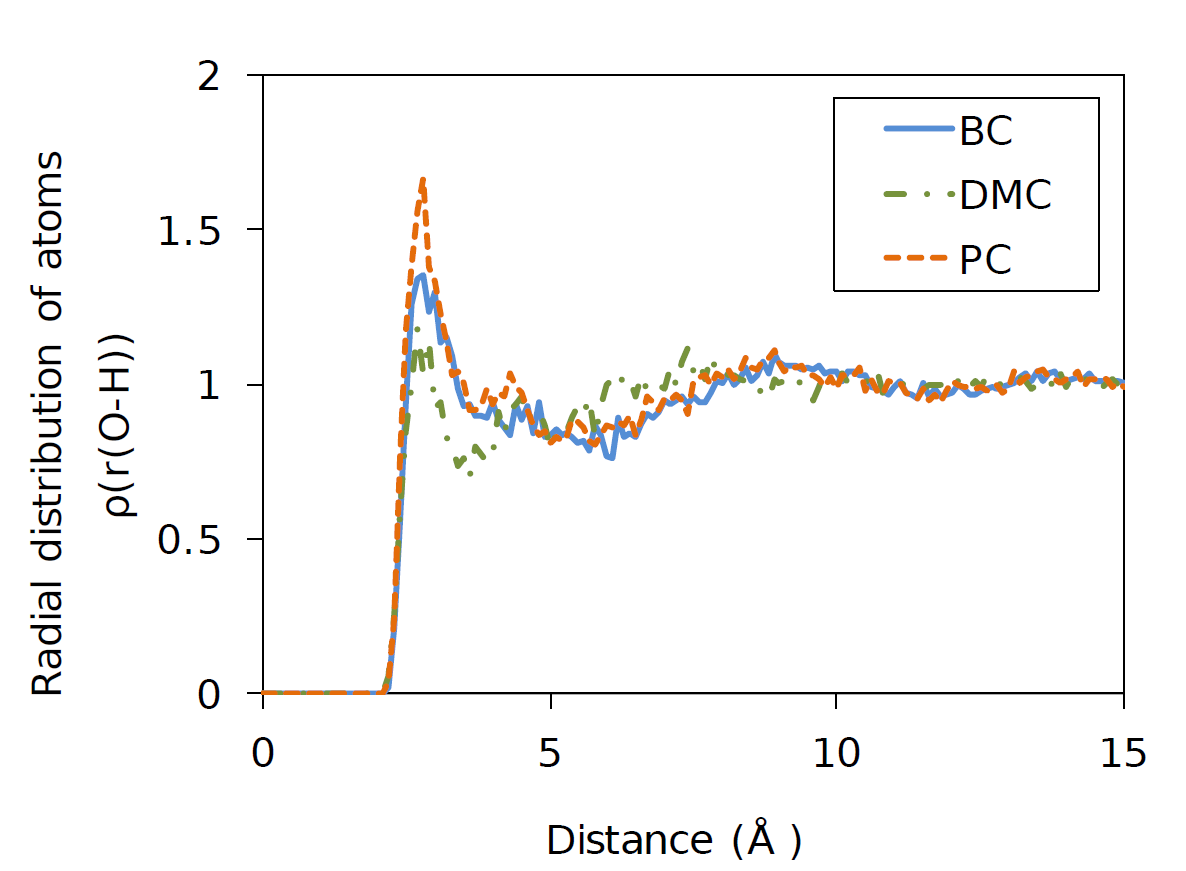
上図はBC、DMCおよびPCにおけるO-H間の分子間距離の動径分布関数です。横軸がO-H間距離、縦軸が1個のO原子を中心に水素原子が何個存在するかという確率密度を表しています。約2.8Å付近にある最も大きいピークは分子間の水素結合に由来しており、各化合物の水素結合の強さを反映しています。この動径分布関数より、DMCは他2つの化合物と比較して水素結合が弱いと考えられますが、一方でDMCのGKcの値はそれらの中で最大の値を示しています。このことは、BCおよびPCは環状分子ですが、DMCが鎖状分子であるため分子間結合ネットワークが形成しやすく、配勾性が強いと考えられます。MDシミュレーションの導入により、水素結合だけでなく、分子形状や嵩高さといったファクターを考慮することで、得られる物性値が改善できることが示されたと言えます。